- (i) A normal healthy cell progressing to a cancerous state.
- (ii) A single cancerous cell progressing to distal metastatic lesions.
Molecular Models for Cancer Progression
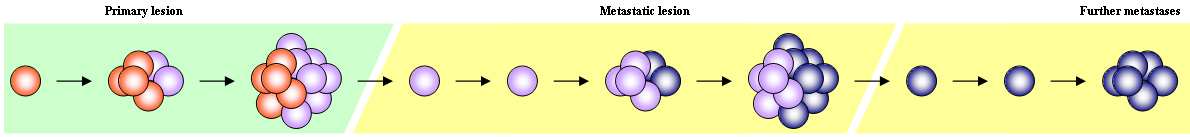 |
Also referred to as somatic evolution, this model rests on the idea that cancer progresses from a weakly metastatic state to a strongly metastatic state through the accumulation of molecular changes (e.g. mutations and/or epigenetic changes) which increase the invasive and proliferative potential of cancer cells. This model fits clinical observations reasonably well (i.e. that melanoma begins as a small easily curable primary lesion and progresses via distinct clinical stages to untreatable metastatic disease) and has been widely accepted for several decades (Miller & Mihm, 2006).
However, this model does not explain the persistent heterogeneity of metastatic melanoma cells (in which so-called progression markers are frequently observed to express inappropriately to clinical stage). Nor does it offer satisfactory explanations for persistent therapy failure.
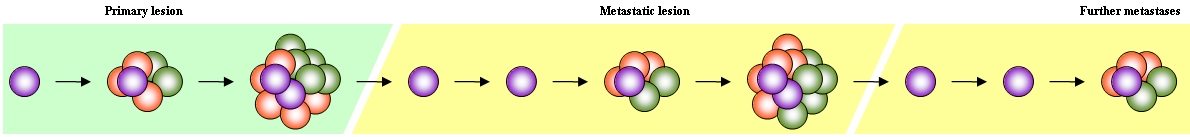 |
This hypothesis evokes the presence of a small population of cells which are solely responsible for propagation of metastases. They also give rise to other cells, but these other cells do not have sufficient replicative potential to drive sustained progression. This model fits observed cancer cell heterogeneity and offers an explanation as to why cancer has evaded treatment. Melanoma stem cells (aka melanoma initiating cells) have been described using NOD/SCID mice (Schatton et al., 2008).
However, while many studies show that CSCs are tumorigenic, nobody has shown that they are especially motile or invasive. So the hypothesis does not address progression particularly well. Furthermore, Sean Morrison's group used NOD/SCID Il2rg-/- mice and single-cell transfection techniques to show that as many as a quarter of all melanoma cells within a given lesion have tumorigenic capacity (Quintana et al., 2008). In addition, other experimenters have shown that the Fc portion of antibodies used in FACS severely reduce in vivo survival of xenografted cells (Taussig et al., 2008). These findings cast serious doubt on the CSC hypothesis for melanoma.
 |
This model, developed by us (Hoek et al., 2006; Hoek et al., 2008a), suggests that cancer cells switch their molecular programming between proliferation and invasion. A central premise is that during proliferation the invasive programming is shut down, and during invasion the proliferative program is shut down. We think that cells switch between programs (phenotypes) in response to microenvironmental cues.
Like the CSC hypothesis, this model fits observed cancer cell heterogeneity, and also offers an explanation as to why cancer has evaded treatment. Furthermore, it explains how the disease progresses to metastasis.